Zdążyć przed Panem Bogiem – omówienie sprawozdania finansowego Mabion po III kwartale 2019 r. Spółka przez wielu obserwatorów nazywana jest spółką jednego projektu. Nie sposób się z tym nie zgodzić, ponieważ obecna wycena jest wypadkową oczekiwań inwestorów w stosunku do projektu Mabion CD20. Jest to lek onkologiczny, biopodobny (ang. biosimilars) do preparatu MabThera/Rituxan, który jest oryginalnym produktem Roche. Stosowany jest on w leczeniu nowotworów krwi oraz reumatoidalnego zapalenia stawów. Na rynkach USA, Japonii i Kanady, preparat sprzedawany jest przez Roche pod nazwą Rituxan, a na pozostałych rynkach objętych sprzedażą lek znany jest pod nazwą MabThera (w tym w Europie). Produkt Roche był trzecim najlepiej sprzedającym się lekiem na całym świecie w 2016 roku. Rynek rytuksymabu warty był wtedy 7,5 mld dolarów. Według danych GlobalData, pierwsze miejsce w rankingu zajęła Humira (producent AbbVie i Eisai, rynek: 16 mld dolarów), a drugie miejsce przypadło Harvoni (producent: Bristol-Myers, rynek: 9,01 mld dolarów). W tym samym rankingu czwarte i piąte miejsce zajęły również leki szwajcarskiego Roche: Avastin i Herceptin.
Rynek USAW 2019 roku upłynęła ochrona patentowa Roche na lek Rituxan w USA – największy rynek farmaceutyczny na świecie. Otworzyło to drogę dla leków generycznych i biopodobnych, które z roku na rok są coraz lepiej postrzegane przez Europejską Agencję Leków (European Medicines Agency – EMA) i jej amerykański odpowiednik (Food and Drug Adminstration – FDA), ze względu na chęć zwiększania dostępności terapii wśród społeczeństwa. W połowie 2018 roku, FDA dopuściła do stosowania na terenie USA lek biopodobny koreańskiej firmy Celltrion – Truxima. Sprzedaż tego leku ruszyła na jesieni tego roku wraz z wygaśnięciem patentu Roche w USA. Oferowany lek jest tańszy o 10 proc. od leku referencyjnego Roche, lecz jego cena z pewnością będzie spadać wraz z wchodzeniem nowych odpowiedników na rynek. Trzeci w kolejce jest Pfizer, którego lek został zatwierdzony w czerwcu, a firma ogłosiła, że sprzedaż rozpocznie na początku 2020 roku. Dodatkowo, kilka dni temu Amgen ogłosił, że złożył aplikację w sprawie zatwierdzenia leku do sprzedaży przez FDA. W związku z tym, jeśli wszystko pójdzie zgodnie z planem, Mabion ze swoim CD20 może wejść na rynek amerykański jako 4 lek biopodobny. W III fazie klinicznej FDA znajduje się także kolejny biosimilars, za którego rozwój odpowiada spółka z grupy Samsung – Archigen Biotech. Według doniesień z października tego roku, ze względu na poślizg spowodowany w badaniach klinicznych, spółka prawdopodobnie nie będzie kontynuowała procesu rejestracji leku, nawet jeśli ostatnia faza badań klinicznych zakończy się powodzeniem. Może mieć to związek z tym, że Samsung doszedł do wniosku, że wejście z kolejnym lekiem biopodobnym do Rituxanu na rynek amerykański nie będzie wystarczająco opłacalne ekonomicznie. Na podobny ruch rok temu zdecydował się Sandoz, który sprzedaje już swój lek biopodobny w Europie. Jako powód podano, że wykonanie dodatkowych badań, jakie wymagała FDA podważa sens ekonomiczny kontynuacji procesu w obliczu przyszłej konkurencji na rynku rituximabu.
kliknij, aby powiększyćŹródło:
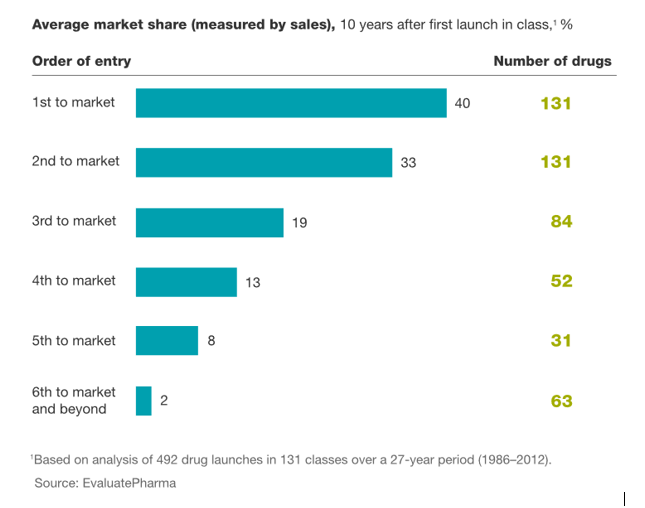
www.mckinsey.com/industries/ph...W kontekście wejścia kolejnego leku na rynek, warto przywołać dane z raportu EvaluePharma z lat 1986-2012. Powyższa tabela pokazuje jaki średni udział rynkowy są w stanie zdobyć po 10 latach kolejne preparaty wchodzące na rynek. Oczywiście największy udział w rynku zawsze będzie miał lek, który wszedł na rynek jako pierwszy. Oczekiwany udział rynkowy dla leku 3 i 4 w kolejce to już jedynie kolejno 19 i 13 proc. W przypadku Mabion CD20 można zakładać, że lek pojawi się jako 5 na amerykańskim rynku (za referencyjnym preparatem Roche, Celltrion, Pfizer i Amgen), co daje mu oczekiwany docelowy udział w rynku na poziomie 8 proc.
Na 22 stycznia 2020 roku, FDA wyznaczyła Mabionowi spotkanie BPD Typu 3 w sprawie rejestracji leku Mabion CD20 na terenie USA. Spotkanie ma na celu m.in. określenie dalszego zakresu prac w kierunku rejestracji leku w USA oraz szczegółów badania pomostowego, które będzie wymagało udowodnienia podobieństwa leku do sprzedawanego w USA Rituxanu. Wymagania dotyczące badania pomostowego zostaną określone przez FDA. W optymistycznym scenariuszu można zakładać, że dodatkowe badanie będzie wymagało przeprowadzeniu badań na 100-200 pacjentach, co może przełożyć się na około rok dodatkowych prac. Z drugiej strony, FDA może zażądać bardziej rozbudowanego badania, co jednocześnie wydłuży całą procedurę i zmniejszy opłacalność projektu amerykańskiego.
Opłacalnością ekonomiczną z pewnością będzie się jednak kierował potencjalny partner Mabionu w USA. Na ten moment spółka ma podpisaną umowę dystrybucyjną z Mylanem w UE. Zgodnie z warunkami partneringu, Mylan ma 30 dni na podpisanie umowy partneringowej na USA do 30 dni po opublikowaniu raportu FDA ze spotkania BPD, które odbędzie się 22 stycznia. Można domniemywać, że sam raport ukaże się około miesiąc po spotkaniu, stąd losy współpracy między Mabionem a Mylanem w USA wyjaśnią się już w kwietniu. Jeśli Mylan nie zdecyduje się w tym czasie na podpisanie partneringu z Mabionem na rynek amerykański, polska spółka ma otwartą drogę do negocjacji z innymi firmami.
Smaczku negocjacjom dodaje fakt, że Mylan połączy się z wydzieloną częścią Pfizera, a nowa spółka będzie działać pod nazwą Upjohn. Pfizer do nowego podmiotu ma nie wnosić projektów związanych z rozwojem leków biopodobnych, a w swojej przyszłej działalności sam Pfizer chce skupić się na rozwoju leków w pełni innowacyjnych. Dodatkowo, obecni akcjonariusze Pfizera (do którego należy biopodobny odpowiednik Rituxanu w USA) nie będą akcjonariuszami Upjohnu, co wyklucza potencjalny konflikt interesów w przypadku podpisaniu przez Mylan umowy partneringowej na USA z Mabionem. Z tego punktu widzenia, połączenie części biznesu Pfizera z Mylanem wydaje się niegroźne dla współpracy z polską spółką. Do myślenia skłania jednak profil działalności Uphjonu, który ma skupiać się na kilku obszarach terapeutycznych: układ sercowo-naczyniowy, psychiatria, urologia i leczenie bólu. Próżno szukać tutaj onkologii, czy immunologii, do którego zaliczane jest reumatoidalne zapalenie stawów. To wraz z długą kolejką chętnych na amerykański rynek może zniechęcać Mylan do podpisania umowy z Mabionem na dystrybucję w USA.
Rynek europejskiOchrona patentowa leku referencyjnego w Europie upłynęła w 2013 roku. To właśnie o rynek europejski obecnie toczy się walka w EMA. Na samym początku warto spojrzeć na wykres poniżej, który pokazuje sprzedaż oryginalnego leku Roche na rynkach w USA, UE, Japonii i pozostałych krajach.
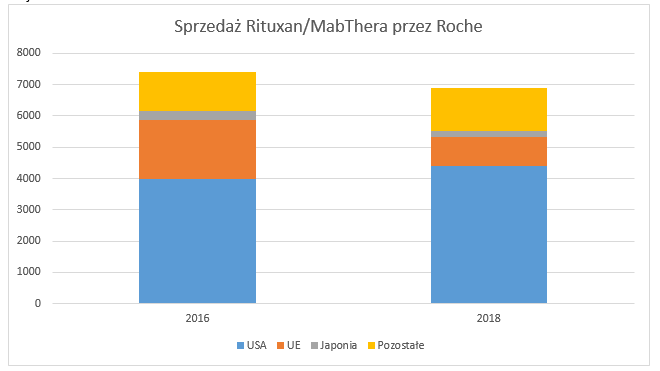
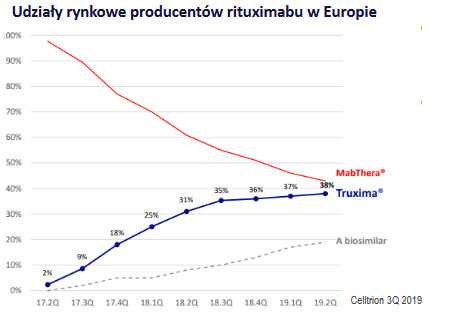
kliknij, aby powiększyćJak widzimy całościowa sprzedaż Rituxanu/MabThera przez Roche spadła w 2018 roku w porównaniu do 2016 roku. Nie miało to jednak nic wspólnego ze spadającym rynkiem, a jedynie wejściem do gry produktów biopodobnych. Choć patent Roche wygasł w UE już w 2013 roku, to do 2 kwartału 2017 roku Roche posiadał monopol na sprzedaż w Europie. W tamtym momencie ze sprzedażą ruszył koreański Celltrion i Sandoz, co przyczyniło się do stopniowego zmniejszania udziału MabThery w Europie. Doprowadziło to do spadku przychodów ze sprzedaży oryginalnego leku Roche z 1,9 mld dolarów w 2016 roku do jedynie 939 mln dolarów w 2018 roku. Ta wartość cały czas spada, a dzieje się to przy stabilnym rynku sprzedaży i znacznie niższych cenach leków biopodobnych Truximy i Sandozu, które z kwartału na kwartał zdobywają europejski rynek rytuksymabu. Leki biopodobne w 2017 roku wchodziły na rynek z cenami niższymi o 30-40 proc. względem leku Roche.
Na rynku europejskim w aktywnej sprzedaży mamy oryginalny produkt Roche, Truximę od Celltrionu oraz preparat Sandozu. W trakcie procedury rejestracyjnej znajduje się także molekula od Pfizer, a w związku z tym, że została ona już wcześniej zatwierdzona przez FDA, prawdopodobieństwo jej oficjalnej rejestracji w UE na początku 2020 roku jest bardzo wysokie. Stąd Mabion w wyścigu o rynek europejski jest również na 5 miejscu. Jeśli weźmiemy pod uwagę, wartości poszczególnych rynków z 2016 roku (dla uproszczenia kalkulacji, ponieważ w tamtym momencie w sprzedaży dostępny był tylko lek referencyjny), to Mabion może celować w kawałek tortu na poziomie 150 mln dolarów (1,9 mld dolarów razy 8 proc.). Oczywiście jest to wartość na 2016 rok. Biorąc pod uwagę, że molekuła Mabionu będzie dopiero 5 na rynku, to do tego czasu wartość całego rynku może spaść, jeśli weźmiemy pod uwagę, że ceny kolejnych preparatów biopodobnych będą coraz niższe. W związku z tym, sprzedaż Mabion CD20 w UE może wynieść nie 150 mln dolarów rocznie, a jedynie 100 mln dolarów.
kliknij, aby powiększyćUmowa z Mylanem zakłada otrzymanie przez spółkę tantiem od przychodów netto ze sprzedaży oraz partycypację w kosztach w stosunku do udziału w przychodach. Zakładając, że średnia marża na sprzedaży w przypadku spółek produkujących leki (np. Roche) to przedział między 30-40 proc., a tantiemy w przypadku podobnych rodzajów umów wynoszą około 40-50 proc. (tantiemy oficjalnie nie zostały opublikowane przez spółkę), to oczekiwany CF powinien wynieść 16 mln dolarów (100 mln dolarów x 35 proc. x 45 proc.). Podobne tantiemy jak i marża na sprzedaży powinna zostać osiągnięta na rynku amerykańskim po podpisania umowy partneringowej, jednak przy dwukrotnie wyższej wartości rynku. Na podstawie tej prostej kalkulacji widać, że rozpoczęcie jak najszybszej sprzedaży w USA jest dużo istotniejsze z perspektywy samej wyceny niż start sprzedaży w UE. Jednak udana procedura rejestracyjna jest bardzo mało prawdopodobna bez akceptacji leku przez EMA w Europie. Dlaczego?
Po pierwsze, FDA dużo bardziej przychylnie będzie spoglądała, jeśli lek będzie zatwierdzony już w UE. W tej kwestii warto jeszcze poruszyć jedną rzecz. Mianowicie, chodzi o to, że w UE lekiem referencyjnym jest MabThera (od Roche) i to właśnie w stosunku do niego Mabion wykazuje podobieństwo swojej molekuły. W USA lekiem dopuszczonym do sprzedaży jest Rituxan (od Roche). Jeśli będzie wiadomo, że Mabion CD20 jest lekiem biopodobnym do MabThery, to dużo łatwiej będzie wykazać to samo do Rituxanu (skoro Rituxan i MabThera to te same preparaty sprzedawane tylko pod inną nazwą).
Po drugie, proces rejestracyjny w FDA jest bardzo kosztowny. Spółce będzie bardzo ciężko pozyskać środki we własnym zakresie. Podpisanie umowy partneringowej w USA, przed rozpoczęciem kosztownej rejestracji za oceanem byłoby dużym ułatwieniem. Wynika to z tego, że umowa wiązałaby się z otrzymaniem płatności wstępnej od partnera (tzw. upfront payment). Umowa z Mylanem w UE zawierała płatność wstępną w wysokości 10 mln dolarów. W przypadku USA, gdzie rynek jest dwukrotnie większy można konserwatywnie oczekiwać 20 mln dolarów. Dlaczego konserwatywnie? Ponieważ przed podpisaniem partneringu na USA, kluczowe będzie dopięcie procesu rejestracyjnego w EMA. Ponownie, skoro molekuła została zaakceptowana przez europejską agencję, to rośnie prawdopodobieństwo, że FDA ją zaakceptuję, co zwiększa potencjalną wartość transakcji. Dodatkowo, podpisanie partneringu w USA i udana rejestracja w UE zwiększyłaby wiarygodność spółki w oczach banków, co pozwoliłoby pozyskać jej finansowanie w postaci dodatkowych kredytów.
Po trzecie, szansę na szybką rejestrację w EMA po grudniowym spotkaniu delikatnie spadły. Z czym ma to związek? Chodzi o skalowalność produkcji. Aby produkcja komercyjna leku miała sens, spółka musi zwiększyć moce produkcyjne, co obecnie trwa. Pytania, które skierowała EMA do spółki dotyczą przede wszystkim procesu komercyjnego wytwarzania Mabion CD20 i podobieństwa produktu w procesie komercyjnym do tego, jaki wytwarzany był w procesie klinicznym. Spółka po spotkaniu poinformowała, że obecnie znajduje się w połowie drogi do dojścia do komercyjnych mocy produkcyjnych (proces może potrwać jeszcze 2-3 miesiące). Wcześniej spółka zakładała, że będzie mogła udowodnić podobieństwo leku w procesie komercyjnym do tego z badania klinicznego już po rejestracji leku, ale przed rozpoczęciem sprzedaży na rynku (spółka mogłaby sprzedawać lek, jednak skala produkcji nie uzasadniałaby ekonomicznego sensu komercjalizacji projektu). Obecnie z mojego punktu widzenia szansę na to maleją, co nie zmienia oczekiwanego momentu rozpoczęcia sprzedaży Mabion CD20 w Europie (2 połowa 2021 roku), jednak przesuwa w czasie moment rejestracji leku, który jest na ten moment kluczowy, aby rozpocząć procedurę w USA (z punktu widzenia finansowania). W listopadowej prezentacji spółki napisano, że EMA potrzebuje około 2-3 kwartałów na ocenę wniosku o zmianę porejestracyjną. Dodatkowo spółka będzie musiała przygotować dossier w kontekście podobieństwa leku komercyjnego do leku z badania klinicznego. Moim zdaniem będzie musiało to nastąpić jeszcze przed oficjalną rejestracją (w innym wypadku, EMA mogła przecież już zatwierdzić lek w grudniu).
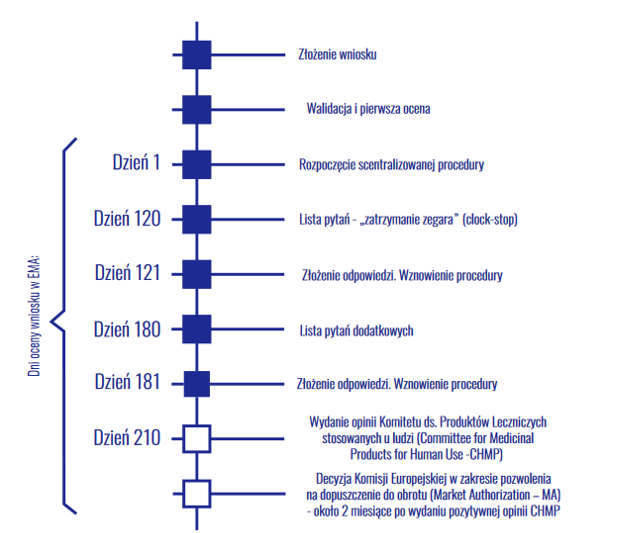
kliknij, aby powiększyćSam schemat procedury wygląda tak jak na powyższej grafice. Grudniowe spotkanie CHMP miało przynieść opinię dotyczącą leku Mabion CD20, co w przypadku pozytywnego komunikatu otwierałoby drogę do rejestracji (około 2 miesiące po opinii CHMP). EMA nie odrzuciła wniosku, jednak wydała dodatkowe zapytania w kontekście podobieństwa leku po przeprowadzeniu procesu zwiększenia mocy produkcyjnych. Wniosek po opublikowaniu dodatkowej serii pytań ponownie cofnął się na etap 180 dnia.
Sytuacja finansowaSpółka nie raportuje żadnych przychodów, chodź na przestrzeni ostatnich lat otrzymała milestona od Mylanu na łączną kwotę 5 mln dolarów z racji osiągnięcia kolejnego kamienia milowego oraz tzw. upfront payment w wysokości 10 mln dolarów. Do otrzymania pozostało 30 mln dolarów (część po otrzymaniu pozytywnej decyzji EMA, kolejna część po rozpoczęciu sprzedaży w Europie). Gdzie pokazywane są otrzymywane płatności od Mylanu, skoro nie ma ich w rachunku zysków i strat?
Kwota otrzymanych milestonów od Mylanu znajduje się po stronie pasywów w pozycji zwrotne zaliczki na poczet praw do dystrybucji oraz przychodach przyszłych okresów. W pierwszej pozycji księgowane są zaliczki, które mogą podlegać zwrotowi i są traktowane przez spółkę jako zobowiązanie bieżące. Kwota milestonów, które zostały otrzymane od Mylanu i mogą podlegać zwrotowi wynosi na koniec 3 kwartału 45 mln złotych. Pozostałe 14 mln złotych znajduje się w przychodach przyszłych okresów i zostanie rozpoznana w rachunku zysków i strat po dopuszczeniu Mabion CD20 do obrotu. Ta część zgodnie z warunkami umowy nie jest już zwrotna. W przychodach przyszłych okresów znajdują się także dotacje na rzeczowe aktywa trwałe oraz prace badawcze w kwocie ponad 30 mln złotych.
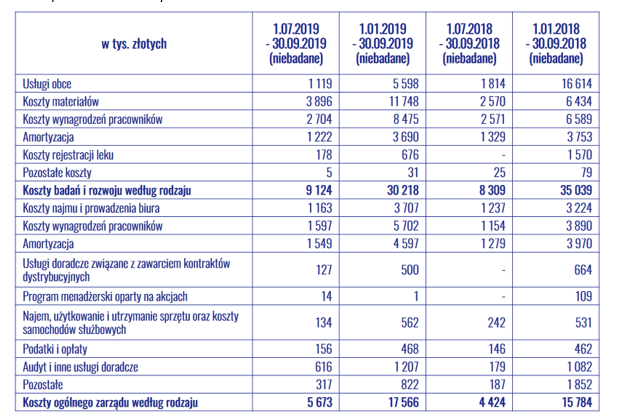
kliknij, aby powiększyćIstotną kwestią z punktu widzenia spółki w obliczu przedłużającej się procedury w EMA jest jej sytuacja płynnościowa. Na koniec III kwartału spółka miała 24 mln złotych środków pieniężnych. Koszty prowadzonych prac badawczych oraz utrzymania spółki wynoszą około 15 mln złotych na kwartał. Z tym stanem gotówki, środków na prowadzenie działalności spółce starczyłoby do końca 2019 roku. Z pomocą przyszedł jednak EBI, który na atrakcyjnych warunkach udzielił spółce finansowania w postaci kredytu na kwotę 30 mln euro. Koszt kredytu to maksymalnie 2,7 proc. W związku z udzieleniem kredytu wyemitowano jednak warranty subskrypcyjne, które będą uprawniały EBI do otrzymania za darmo akcji nowej emisji akcji. W związku z tym NWZA na początku grudnia przegłosowało nową emisję akcji, która zwiększy ich ilość o 2,85 proc. Rozwodnienie nie jest duże, a może być jeszcze mniejsze, biorąc pod uwagę fakt, że kredyt w wysokości 30 mln euro będzie udostępniany w transzach. W związku z tym, można domniemywać, że warranty będą otrzymywane przez EBI w transzach proporcjonalnie do wydanych środków spółce w ramach finansowania. W tym wypadku słowo transza ma bardzo duże znaczenie, ponieważ nie wiadomo jaka część kwoty została udostępniona spółce na ten moment, a ile z tego było warunkowane uzyskaniem pozytywnej opinii EMA. Obecnie pozostaje to kluczową zagadką.
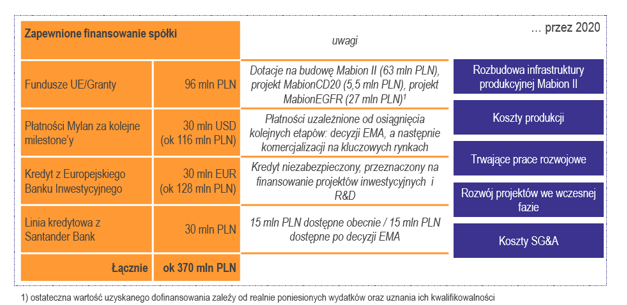
kliknij, aby powiększyćW slajdzie zaprezentowanym na listopadowej prezentacji pokazano, że zapewniono finansowanie spółki w kwocie 370 mln złotych. Znaczna część tych kwot jest jednak warunkowa (płatności od Mylan zależą od decyzji EMA, 15 mln złotych z Santanderu warunkowane jest pozytywną decyzją EMA, podobnie z kredytem z EBI, którego szczegółowe warunki pozostają niewiadomą). Fundusze UE/Granty są zazwyczaj otrzymywane dopiero po poniesieniu kosztów przez spółkę, więc najpierw te środki spółka musi mieć. Budowa fabryki Mabion II potrzebna jest do tego, aby sprostać wolumenom produkcyjnym w przypadku rozpoczęcia sprzedaży w USA. Całkowity koszt inwestycji w Mabion II szacowany jest na 170 mln złotych (110 mln złotych do pokrycia z własnego finansowania), a przewidywany czas zakończenia inwestycji w umowie określony był na koniec 2021 roku. W tym momencie budowa jeszcze się nie rozpoczęła, co stwarza ryzyko utraty tego finansowania w przypadku dalszego przedłużania się procedury w EMA.
Z punktu widzenia operacyjnego również zbyt dużo nie można powiedzieć o wynikach spółki, które są nie istotne z punktu widzenia kursu spółki aż do momentu rozpoczęcia sprzedaży leku na którymś z rynków. Zysk EBIT od wielu kwartałów odzwierciedla ponoszone tylko i wyłącznie koszty operacyjne, które w spółce wahają się w przedziale 10-20 mln złotych na kwartał.
Podsumowanie:Najważniejszą kwestią dla spółki jest uzyskanie pozytywnej decyzji EMA jak najszybciej. Pozwoliłoby to uzyskać kolejną płatność w ramach kamienia milowego od Mylanu oraz z dużo większym optymizmem patrzyć na rozpoczynającą się procedurę w FDA. Dużym ryzykiem jest jednak możliwość przesunięcia samej decyzji o kolejne kilka kwartałów, co pozostawiłoby spółkę w bardzo trudnej sytuacji. Do prowadzenia obecnej działalności spółka potrzebuje 15 mln złotych na kwartał. Środki pieniężne na koniec 3 kwartału wynosiły 24 mln złotych, co w połączeniu z dostępnym obecnie limitem w Santanderze w wysokości 15 mln złotych, dają spółce względny spokój do końca marca. Pytaniem pozostaje to ile kredytu zostało udostępnione na start w ramach umowy z EBI. Zakładając, że było to 10 mln euro, to spółka wydaje się mieć zapewnione finansowanie do końca 2020 roku przy takich samych kosztach działalności jak teraz. Nie pozwoli to jednak na rozpoczęcie badania pomostowego w USA, czy rozszerzenia prac w prowadzonych równolegle projektach takich jak Mabion EFGR, czy Mabion CD20 w stwardnieniu rozsianym. Stwarza to również ryzyko utraty dotacji na Mabion II. Potwierdza to tezę, że zarówno obecna działalność, wejście nowych projektów do portfolio spółki, czy rejestracja w USA jest uzależniona w dużym stopniu od pozytywnej decyzji EMA, której wciąż nie ma. Dużym ułatwieniem dla Mabionu byłaby zgoda EMA na wprowadzenie zmian porejestracyjnych związanych ze skalowalnością produkcji już po zatwierdzeniu leku, co było oczekiwane jeszcze w listopadzie. Obecnie jednak wydaje się, że szansę na to zmalały, co zwiększa ryzyko dla spółki w kontekście finansowania dalszej działalności. Z drugiej strony pozytywna i szybka decyzja otwiera ogromne możliwości przed spółką, przede wszystkim w kontekście dalszego finansowania, jak i zwiększa w moim oczach szansę na wejście na bardzo lukratywny rynek w USA.
Powyższa treść przez 365 dni była zarezerwowana tylko dla osób posiadających abonament.