Proces opracowywania i udostępnienia pacjentom nowego leku jest bardzo długi, może trwać od 8 do nawet 17 lat.
Proces odkrywania, rozwoju i produkcji leku nie jest ani łatwy, ani tym bardziej szybki. Proces ten potrafi zająć nawet kilkanaście lat, a przypadki takie jak obecnie (szczepionka przeciwko Covid-19) są w zdecydowanej części wyjątkami niż regułą. Proszę zresztą spojrzeć na poniższe wykresy ukazujące proces odkrywania i rozwoju leków w czasie.
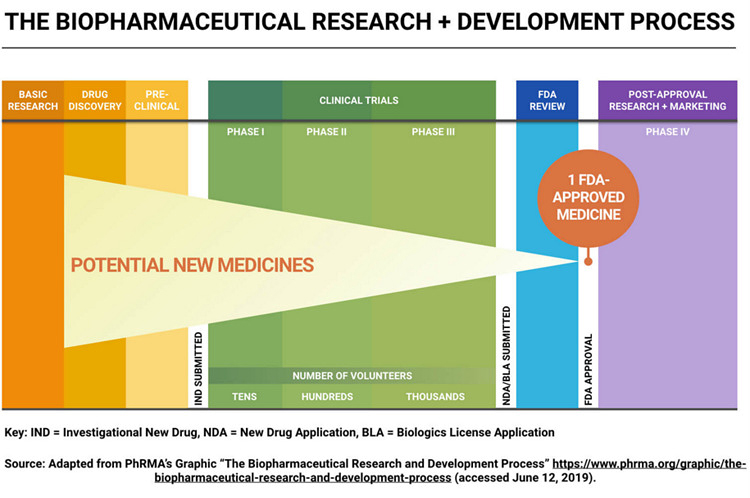
Badania biofarmaceutyczne oraz proces rozwoju leku
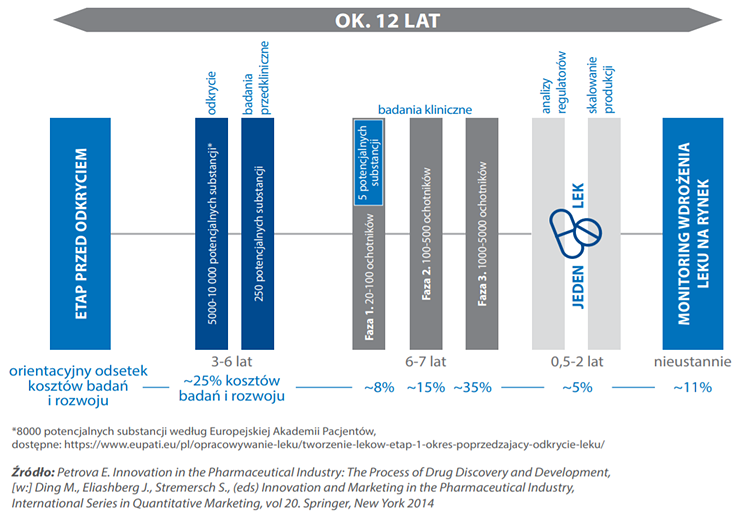
Proces opracowywania i udostępniania pacjentom nowego leku.
Co ważne, wszystkie wykresy i dane przedstawione w tej serii artykułów należy traktować jako informacje przykładowe i nie należy się specjalnie przywiązywać do podawanych wartości, ponieważ są one w dużej mierze orientacyjnie. Tym niemniej już tutaj widać, że potrzebujemy po prostu wielu prób, wielu badań i dużo czasu, by na końcu otrzymać jeden dopuszczony do obrotu lek. Z każdym kolejnym etapem w proces odkrywania leku będą odpadać kolejne cząsteczki (kandydaci na lek), które po prostu nie spełniają wyśrubowanych na szczęście kryteriów. W literaturze przedmiotu przyjmuje się, że trzeba przebadać początkowo kilka tysięcy związków (w pierwszym momencie w fazie odkrywania – drug discovery), aby w efekcie na samym końcu odnieść sukces. A przypomnę na podstawie poprzedniego artykułu z serii, że rocznie zatwierdzanych jest w USA zaledwie kilkadziesiąt nowych leków.
Na przedstawionych poniżej grafikach (pochodzą z prezentacji Pure Biologics oraz Onco Arendi Therapeutics) widać cykl rozwoju leków innowacyjnych. Przypomnę, że pierwsza z tych spółek skupia się na rozwoju cząsteczek biologicznych, a druga chemicznych.
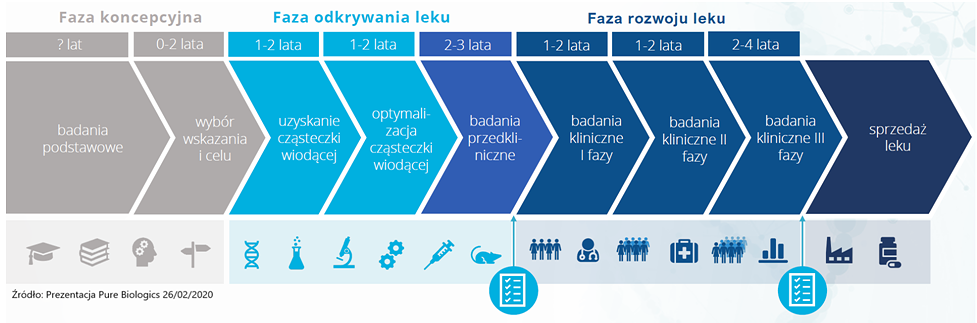
Źródło: Prezentacja Pure Biologics. Kliknij, aby powiększyć.
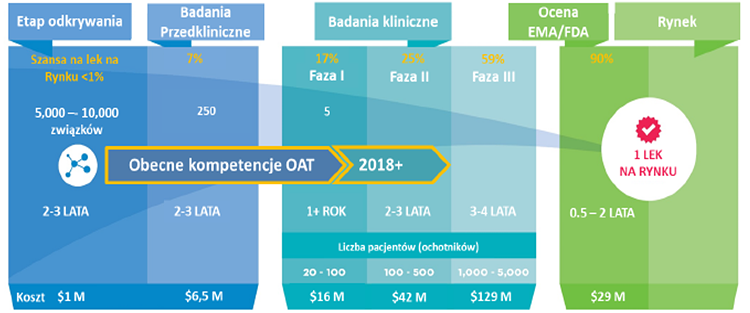
Źródło: Prospekt emisyjny Onco Arendi Therapeutics
Patrząc na powyższe wykresy można oszacować, że proces opracowywania i udostępnienia pacjentom nowego leku może zawierać się w przedziale 8-17 lat (szacunki Pure Biologics) jak i 11-16 lat (informacje z prospektu emisyjnego Onco Arendi Therapeutics). Już tutaj same dolne i górne przedziały tych zakresów pokazują dość dużą (co najmniej kilkudziesięcioprocentową) rozpiętość okresów czasowych. Jeśli więc średnio przyjmiemy, że proces wynalezienia i wprowadzenia na rynek innowacyjnego leku trwa zazwyczaj powyżej 10 lat, to nie będziemy przesadnie pesymistyczni. Warto to zapamiętać, w szczególności jeśli odniesiemy to do kamieni milowych przy komercjalizacji projektów, których znaczną część będzie można otrzymać w ostatnich fazach tego całego procesu.
Pierwsza znacząca umowa partneringowa wśród polskich giełdowych biotechów miała miejsce w marcu 2017 roku i dotyczyła cząsteczki SEL24 w transakcji Selvity (obecnie Ryvu Therapeutics) z podmiotem z włoskiej grupy Menarini. Cząsteczka ta została skomercjalizowana w momencie rozpoczęcia pierwszej fazy badań klinicznych. Natomiast Selvita pozyskała ten projekt w 2008 roku od Fundacji Rozwoju Diagnostyki i Terapii w Warszawie. W chwili obecnej cząsteczka jest w drugiej fazie badan klinicznych. Widzimy więc, że minęło ponad 12 lat, a wciąż mamy jeszcze kilka etapów, aby cząsteczka została pełnoprawnym lekiem. Ten przykład wybitnie pokazuje, że projekty biotechnologiczne charakteryzują się długim lub wręcz bardzo długim okresem czasu.
Co ważne, konkretna cząsteczka, czyli potencjalny lek, może się zachowywać zupełnie inaczej, jeśli chodzi o tempo przechodzenia przez kolejne fazy tego procesu. Musimy, jako inwestorzy, o tym pamiętać, ponieważ będziemy otrzymywali od spółek informacje dotyczące harmonogramów i przewidywanych czasów trwania poszczególnych etapów. Trzeba mieć świadomość, że są to terminy bardzo szacunkowe i przybliżone, a czasami wręcz optymistyczne. Spółki jednak już coraz częściej zabezpieczają się i w prezentowanych informacjach czy prezentacjach umieszczają disclaimery: „Termin realizacji podany jest w przybliżeniu i szacowany na podstawie wyników badań i rozwoju. Specyfika eksperymentalnych badań naukowych, przemysłowych i prac rozwojowych sprawia, że w przyszłości terminy te mogą ulec istotnej zmianie.” Jest to oczywiście pewne zabezpieczenie prawne, ale również szczera prawda – w biotechnologii nic nie jest pewne, zwłaszcza terminy.
Warto jeszcze dodać, że powyższe wykresy należy przede wszystkim odnosić do procesu odkrywania i wytwarzania leków innowacyjnych. Leki biopodobne oraz szczególnie generyczne leki chemiczne mają trochę prostszą drogę. W wypadku chemicznych leków generycznych zazwyczaj producent takiego zamiennika nie musi już wykonywać zaawansowanych i drogich badań przedklinicznych i klinicznych, ponieważ wykorzystuje się wyniki badań wykonane dla leku innowacyjnego. W wypadku leków biopodobnych jest trochę trudniej, ze względu na ich skomplikowanie i brak możliwości odwzorowania w 100 procentach składu leku innowacyjnego. W przypadku tych leków do każdego zgłoszenia podchodzi się indywidualnie, tym niemniej można przyjąć, że jednak sam proces (a co za tym idzie czas i koszty) zazwyczaj jest także krótszy niż przy rejestracji innowacyjnego leku biologicznego.
Etapy
Odkrywanie leków i późniejsze fazy procesu są dość skomplikowane, a poniżej grafika prezentująca poszczególne etapy.
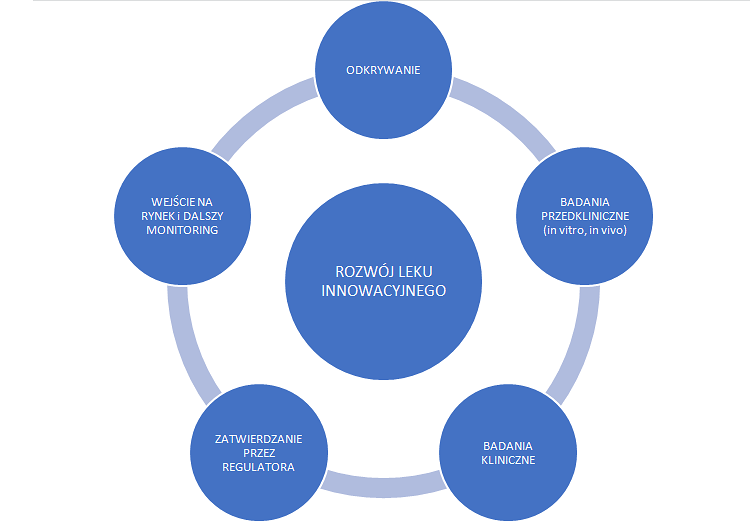
Na początku mamy etap odkrywania leku, który często zabiera wiele miesięcy. Jest tutaj zarówno faza koncepcyjna, zwana inaczej fazą poszukiwań i hipotez badawczych, jak i sama faza odkrywania. W zasadzie w każdej branży opracowywanie nowego produktu rozpoczynamy od analizy rynku i poszukiwania niezaspokojonych potrzeb. W przypadku biznesu farmaceutycznego takimi potrzebami są w praktyce jednostki chorobowe, na które albo nie ma skutecznego leku albo dostępne terapie powodują dotkliwe działania niepożądane. Oznacza to, że na początku trzeba zrozumieć mechanizm danej choroby i poszukać sposobu, dzięki któremu da się ją cofnąć lub przynajmniej zahamować. To oczywiście wymaga podjęcia wielu prób, często zakończonych niepowodzeniem i właśnie na tych etapach odpada najwięcej cząsteczek i projektów. Warto o tym pamiętać, ponieważ część polskich spółek biotechnologicznych ma projekty na bardzo wczesnym etapie rozwoju. Nierzadko pozyskano na nie nawet finansowanie z NCBiR lub innych agencji. Nie oznacza to jednak, że są to już projekty rozwojowe. Tak naprawdę są to badania, z których większość wcale nie musi zakończyć się pierwszym sukcesem, czyli przejściem do fazy badań przedklinicznych. W zasadzie na tym etapie zazwyczaj dokonuje się ostateczna selekcja cząsteczek wiodących, z których wybiera się najlepszych kandydatów na leki, które przechodzą do kolejnego etapu.
Etap badań przedklinicznych obejmuje działania, które są łącznikiem miedzy odkrywaniem leków a rozpoczęciem badań klinicznych na ludziach. Badania przedkliniczne skupione są na ocenie oddziaływania cząsteczki na komórki/tkanki ludzkie (badania in vitro) oraz zwierzęta (badania in vivo). Ten etap ma na celu wstępną ocenę bezpieczeństwa oraz efektywności kandydata na lek zanim zostanie on pierwszy raz podany ludziom. Tutaj też pozyskuje się informację na temat tego, jak kandydat na lek zachowuje się w organizmie zwierzęcym pod względem absorpcji, metabolizmu czy wydalania. Badania przedkliniczne to częsty moment w czasie, gdy spółka decydują się na patentowanie, aby zacząć prawnie chronić swoje odkrycie. O patentach i sposobach dodatkowej ochrony już w okresie sprzedaży rynkowej leku pisałem w poprzednim artykule. Badania są przeprowadzane zgodnie z dobrą praktyką laboratoryjną (GLP), w ściśle kontrolowanych warunkach. Uzyskane informacje są decydujące, czy cząsteczka zostanie dopuszczona do badań klinicznych – z udziałem zdrowych ochotników. Ten etap rozwoju leku innowacyjnego pomaga również naukowcom w projektowaniu proponowanych badań klinicznych pierwszej fazy na ludziach jak choćby określenie proponowanej dawki początkowej.
Najpierw jednak musimy uzyskać zgodę regulatora na rozpoczęcia badań klinicznych, co np. jeśli chodzi o „najgorętszy” rynek amerykański przejawia się w złożeniu wniosku IND (Investigational New Drug) do FDA (Amerykańska Agencja Żywności i Leków). Wniosek ten musi zawierać m.in. wyniki badań na zwierzętach, informacje na temat produkcji leku, plan przeprowadzenia testów na ludziach. W tym wypadku ważnym jest ustalenie, czy produkt jest wystarczająco bezpieczny do początkowego stosowania u ludzi oraz czy związek wykazuje aktywność farmakologiczną uzasadniającą komercyjny rozwój. Co ważne badania te powinny wykazać, że produkt nie narazi ludzi na nieuzasadnione ryzyko, jeśli będzie stosowany w ograniczonych badaniach klinicznych na wczesnym etapie. Jeśli wniosek IND zostanie zatwierdzony, spółka może przejść do następnego etapu.
Badania kliniczne, trzeba to wyraźnie powiedzieć, są to de facto eksperymenty biologiczne, których uczestnikiem jest człowiek. Oczywiście słowo eksperyment nie powinno być tutaj odbierane pejoratywnie, ponieważ istnieje wiele zabezpieczeń przed podjęciem zbyt dużego ryzyka na tym etapie całego procesu. Badania kliniczne prowadzi się w celu potwierdzenia skutków działania leku ewentualnie w celu zidentyfikowania działań niepożądanych oraz poznania wchłaniania, metabolizmu i wydalania potencjalnych leków. Ogólnie rzecz biorąc, w badaniach klinicznych ocenia się bezpieczeństwo oraz skuteczność kandydatów na leki.
Przechodząc do praktycznego przykładu wiemy, że w trakcie projektu SEL24 opracowano ponad 1.300 związków, z czego prawie 800 zostało zsyntetyzowanych, kilkadziesiąt poddano badaniom in vivo, a tylko jeden został wybrany jako kandydat do badań klinicznych. Tego typu dane pokazują pewną skalę trudności, z jakimi muszą się mierzyć naukowcy chcący wynaleźć nowy lek, a w zasadzie kandydata na lek. I tutaj pewna wskazówka dla inwestorów. W wypadku spółek biotechnologicznych informacja o zatrzymaniu projektu i zaprzestaniu jego kontynuacji może być w pewnych okolicznościach dobrą informacją! Oczywiście na pozór brzmi to dość nielogicznie, aby nie napisać niedorzecznie. Ale proszę spojrzeć na to z innej strony. Decyzja o zamknięciu projektu, który nie jest obiecujący w efekcie przekłada się na zatrzymanie wydawania na niego pieniędzy oraz poświęcania cennego czasu i uwagi naukowców. Pokazuje to też zdolność zarządu do podejmowania twardych, choć nie zawsze popularnych, decyzji. W tym momencie przypomnę, że Ryvu Therapeutics ogłosiło w drugiej połowie 2020 roku zamknięcie dwóch projektów i przeznaczenie środków na kontynuację tych najbardziej obiecujących. Naturalnie wywołało to spadek kursu akcji, ale być może w długim terminie wyjdzie to spółce na dobre. Przecież statystyka dotycząca szans powodzenia projektów jest nieubłagana, o czym zresztą będzie druga część tego artykułu.
Uczciwie trzeba też powiedzieć, że od kilku lat daje się zauważyć coraz większe wykorzystanie sztucznej inteligencji oraz big data w procesie odkrywania leków. To powinno mieć przełożenie na skrócenie okresu trwania etapu odkrywania oraz większego niż wcześniej prawdopodobieństwa skonstruowania takich związków, które mają szansę stania się lekami w przyszłości. A to zaś oznacza, że możemy mieć nadzieję, że już w niedalekiej przyszłości szanse na wynalezienie skutecznych leków (a dla inwestorów dużych pieniędzy) będą rosły.
Inwestorzy czasami zapominają, że dopuszczenie do badań (niezależnie do której fazy) a samo ich rozpoczęcie to niekoniecznie ten sam moment w czasie. Trzeba przecież wynegocjować i podpisać umowy ze szpitalami, zrekrutować pacjentów, wyprodukować i przekazać leki – to wszystko zajmuje czas. Dla przykładu Selvita w kwietniu 2016 roku złożyła zgłoszenie NDA dotyczące związku SEL24 i planowała wtedy rozpocząć badania kliniczne w trzecim kwartale 2016 roku. Koniec końców FDA wydała zgodę na rozpoczęcie badań po czterech miesiącach w sierpniu 2016 roku, a same badania rozpoczęły się w marcu 2017 roku. Z kolei gdybyśmy popatrzyli na kolejną cząsteczkę SEL120, to w marcu 2019 roku spółka uzyskała zgodę FDA na rozpoczęcia badań klinicznych, a pierwsze podanie pacjentowi nastąpiło 6 miesięcy później. Gdy jednak spojrzymy w prezentację spółki z marca 2016 r., to wtedy spółka zakładała, że pierwsza faza ruszy w drugiej połowie 2017 roku.
I faza badań klinicznych przeprowadzana jest zazwyczaj na kilkudziesięciu ochotnikach i jest to pierwszy kontakt organizmu człowieka z nowym lekiem. Celem tej fazy jest wstępne określenie bezpieczeństwa leku oraz wstępnego podawania – minimalne i maksymalne dawki. Generalnie w tej fazie ochotnikami są osoby zdrowe. Wyjątkiem są leki onkologiczne lub inne silnie trujące, ze względu na kwestie etyczne i zazwyczaj ciężkie skutki uboczne proponowanych terapii. W takim wypadku już w pierwszej fazie badań klinicznych podawane są one pacjentom, czytaj osobom chorym. Badania I fazy trwają od „dużych” kilku miesięcy do w zasadzie nieskończoności. Oczywiście tutaj trochę przesadzam, ale znów odwołując się do cząsteczki SEL24, gdzie badania tej fazy trwały 3 lata. Uczciwie trzeba przyznać, że w międzyczasie doszło do partneringu i przekazywania projektu jak i wstrzymanie badań w związku ze śmiercią pacjenta, co zapewne wpłynęło na opóźnienie w harmonogramie projektu. Tym niemniej raz jeszcze podkreślę, że dość często okres rzeczywistego trwania badań klinicznych (nie tylko w pierwszej fazie) jest dłuższy niż ten planowany.
Zakładając, że pierwsza część badań zakończy się sukcesem spółka zgłasza wniosek o rozpoczęcie drugiej fazy, w trakcie której kandydat na lek jest testowany na większej populacji – zazwyczaj do kilkuset pacjentów – czyli osób cierpiących na daną chorobę. Jest to etap niezwykle ważny, gdyż lek podawany jest po raz pierwszy (oprócz onkologii jak wspomniano wcześniej) osobom z daną jednostką chorobową. Celem tej fazy jest określenie dawki (ewentualnie doprecyzowanie) leku, częstotliwości podawania oraz faktycznej skuteczności leczenia. Już sam taki opis może wskazywać na to, że to właśnie ten etap może charakteryzować się znacznym ryzykiem. Jest tak rzeczywiście (do tego jeszcze powrócimy) głównie ze względu na konieczność potwierdzenia faktycznej skuteczności leczenia na ludziach. Badania kliniczne na zwierzętach, nawet gdy wypadają bardzo pozytywnie nie są żadnym gwarantem co do pomyślnego zakończenia procesu wynalezienia skutecznego leku. Musimy pamiętać, że zwierzęta często reagują inaczej niż ludzie na daną substancję leczniczą i w zasadzie nie istnieje model zwierzęcy, który mógłby w pełni umożliwić zastąpienie eksperymentalnego podawania leków ludziom, czyli badań klinicznych.
Stosuje się tutaj porównanie testowanego kandydata na lek z placebo zazwyczaj za pomocą tzw. podwójnej ślepej próby. Oznacza to, że ani pacjent, ani lekarz nie wiedzą, jaka substancja jest podawana choremu, aby oczekiwania tych dwóch stron nie wpływały na wyniki badań. Wykazanie zakładanej skuteczności po przeprowadzonych testach statystycznych i upewnieniu się, że stosunek korzyści do ryzyka jest korzystny, umożliwia dopuszczenie substancji do kolejnej, trzeciej fazy badań klinicznych.
W wypadku drugiej fazy najczęściej przyjmuje się, że zabiera ona ok. 1-2 lat. Patrząc na esketaminę od Celon Pharmy w depresji lekoopornej dwubiegunowej, to II faza (od momentu złożenia wniosku do otrzymania pozytywnych wyników) zajęła 2,5 roku, podczas gdy spółka pierwotnie zakładała, że zamknie się ona w 12 miesiącach Tak naprawdę potrwa to jeszcze chwilę, ponieważ na początku stycznia bieżącego roku spółka ogłosiła, że spodziewa się pełnych danych klinicznych, łącznie z wynikami 6-tygodniowej, dodatkowej obserwacji w ciągu najbliższych 8-12 tygodni.
Trzecia faza badań klinicznych jest tą, gdzie potwierdza się działanie leku na dość dużej populacji od kilkuset do nawet kilku tysięcy chorych. Populacja badawcza powinna być zbliżona do populacji chorych występujących w naturalnym środowisku. Oznacza to, że znaleźć się w niej powinny osoby różnych ras, płci, o różnym stanie zdrowia i stopnia zaawansowania choroby. Faza ta zazwyczaj odbywa się w wielu ośrodkach klinicznych, często w różnych krajach. Tutaj również sprawdza się, czy dany lek jest skuteczniejszy od dotychczas stosowanych terapii, o ile oczywiście takie istnieją. Ta część badań może trwać od roku do kilku lat. Badania tej fazy zbierają dane, które są podstawą do rejestracji (faza III a) oraz służą celom marketingowym (faza III b). W wypadku tej fazy badania często trwają najdłużej, co jest oczywiście w sporej mierze efektem dość rozległej populacji i współpracy z wieloma ośrodkami klinicznymi.
Warto pamiętać, że również w trzeciej fazie badań klinicznych istnieje całkiem spore ryzyko, że lek nie zakończy jej z sukcesem. Dzieje się tak mimo tego, że przecież w drugiej fazie badań taki lek wykazywał ogólne oznaki skuteczności. Proszę jednak pamiętać, że zakres i charakter badań był wtedy ograniczony w porównaniu do fazy trzeciej.
Tutaj jeszcze jedno spostrzeżenie natury ogólnej. Zazwyczaj spółki marzą o uzyskaniu zatwierdzenia przez FDA, co może dać dostęp do największego rynku amerykańskiego. To zaś oznacza, że badania kliniczne zazwyczaj będą w dużej części przeprowadzane w USA. Po pierwsze, ze względu na sam prestiż, po drugie na większą wiarygodność tych badań w oczach FDA. To zaś oznacza, że trzeba być przygotowanym zarówno na wyższe koszty niż np. w Europie, jak i dysponować siatką kontaktów na tym rynku.
Aby nowy lek trafił do normalnej sprzedaży, po potwierdzeniu badaniami klinicznymi skuteczności terapeutycznej i bezpieczeństwa, musi przejść jeszcze długi i skomplikowany proces rejestracji. Na podkreślenie zasługuje fakt, że to spółka wnioskująca o dopuszczenie preparatu na rynek odpowiedzialna jest za przeprowadzenie badań (wstępne, kliniczne i przedkliniczne) i przekazanie dokumentacji. FDA, ale również inni regulatorzy, na podstawie tej dokumentacji wydają swoje decyzje. W wypadku amerykańskiej agencji wiąże się to ze złożeniem dokumentu NDA (New Drug Application). Jest to bardzo szeroka dokumentacja (przekraczająca często nawet kilkanaście-kilkadziesiąt tysięcy stron) opisująca dokładnie cały proces przebiegu powstawania leku, zawierająca wszystkie niezbędne dane z badań zarówno przed jak i klinicznych. Celem jest wykazanie, że lek jest skuteczny, bezpieczny oraz posiada odpowiednią jakość. W skład tej dokumentacji wchodzą również takie rzeczy jak informacja o patentach, skutkach ubocznych, czy proponowana instrukcja stosowania leku.
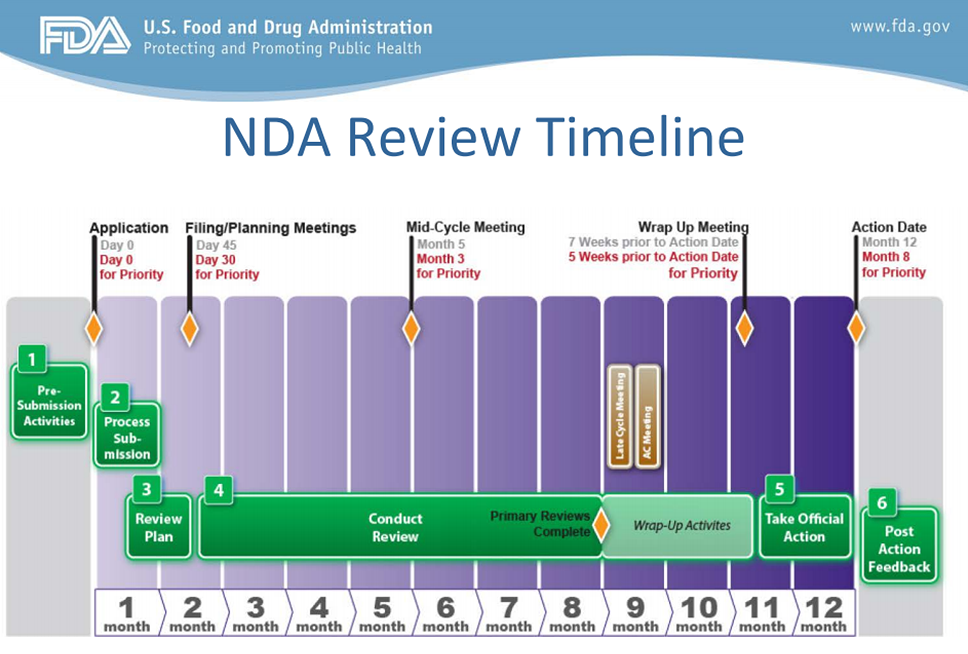
Kliknij, aby powiększyć.
FDA dokładnie przegląda tę dokumentację początkowo pod kątem kompletności. Jeśli na tym etapie wszystko wygląda prawidłowo, to zespół zatwierdzający ma od 6 („priority review”, „Accelerated Approval”, „Breakthrough Therapy” czy „Fast Track”) do 10 miesięcy (standardowa procedura) czasu na zatwierdzenie leku. Przyspieszona ścieżka może być wykorzystana np. przy kandydatach na lek, którzy wykazują istotną poprawę w bezpieczeństwie lub skuteczności w porównaniu z istniejącymi terapiami. Tutaj znowu można wrócić do przypadku szczepionki, której czas zatwierdzenia liczył się de facto w tygodniach, a nie w miesiącach. Można oczywiście także zacząć się zastanawiać w tym kontekście nad cząsteczką OATD-01, która została sprzedana w umowie partneringowej Onco Arendi Therapeutics i Galapagos. Jeśli faktycznie miałaby szansę stać się lekiem leczącym powikłania po Covid-19 (idiopatyczne zwłóknienie płuc), to pojawiłaby się zapewne szansa na skorzystanie z szybszych ścieżek akceptacji, choć nie oczekiwałbym aż takiego przyspieszenia jak w wypadku szczepionek.
Zespół FDA może również poprosić o odpowiedzi na pytanie na podstawie dostępnych danych lub nawet zdecydować o konieczności przeprowadzenia np. dodatkowych badań. Jako inwestorzy powinniśmy pamiętać, że jest to również istotny moment i zdarza się, że spółki wnioskujące wycofują wniosek na tym właśnie etapie, bądź muszą się cofnąć i przeprowadzić dodatkowe badania. Tutaj dobrym przykładem jest lek Mabion CD20, który mając problemy z uzyskaniem zgody EMA dla MabionCD20 w marcu 2020 roku wycofał wniosek o zgodę złożony w EMA (Europejska Agencja Leków). W efekcie musi przeprowadzić dodatkowe badanie fazy I/II, co wydłuży projektu zapewne o 2 lata i oczywiście podniesie istotnie koszty.
Po rejestracji oraz rozpoczęciu sprzedaży nadal istnieje konieczność raportowania tzw. IV fazy badań klinicznych. Celem jest tutaj upewnienie się, że lek jest bezpieczny nie tylko dla wszystkich chorych, którzy go przyjmują, ale również we wszystkich wskazaniach zalecanych przez producenta. Faza ta w naturalny sposób jest rzeczywistym, kolejnym weryfikatorem wcześniej uzyskanych wyników. W tym momencie także zbiera się i dane dotyczące częstotliwości i nasilenia skutków ubocznych a także interakcji z innymi przyjmowanymi przez pacjentów lekami.
Podsumowując, widać, że proces stworzenia innowacyjnego leku jest po prostu bardzo długi. Oczywiście można tutaj powoływać się na szczepionki pandemiczne przeciw COVID-19, które w zasadzie zostały wynalezione i zatwierdzone w okresie poniżej jednego roku. Ja jednak traktowałbym to jako odstępstwo od pewnych norm niż standard, który miałby być użyty np. w wypadku szacowania czasu potrzebnego dla wynalezienia leków przez polskie biotechy. Pandemia to sytuacja szczególna, miejmy nadzieję, że wyjątkowa stąd też była tutaj wyjątkowa mobilizacja po stronie producentów jak i regulatorów. Wspomnę tutaj tylko o przyspieszeniu procesu ze względu na prowadzenie części etapów prac równolegle, zainwestowania ogromnych środków (oraz pracy ludzkiej) czy też wręcz produkcji szczepionki na skalę przemysłową jeszcze przed jej formalnym zatwierdzeniem przez regulatorów. Również zatwierdzający uruchomili specjalne szybkie ścieżki oceny i rozpoczęli analizowanie i ocenianie szczepionki podczas prowadzonych badań klinicznych.
Prawdopodobieństwo – słowo klucz w biotechnologii
W pierwszej części artykułu opisywałem różne fazy pracy nad lekami innowacyjnymi w kontekście potrzebnego na to czasu i wskazywałem na możliwości och wydłużenia. To oczywiste ryzyko dla inwestora (czy i idące za tym pieniądze), które powinien brać pod uwagę. Nie można również zapomnieć o drugim czyli prawdopodobieństwie osiągnięcia sukcesu bądź porażki w każdym z etapów. Proszę spojrzeć na dwa poniższe slajdy.
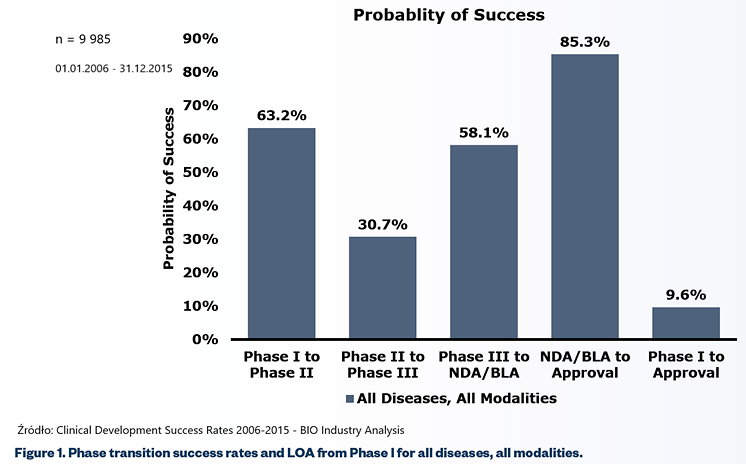
Średnie prawdopodobieństwo przejścia przez kolejne fazy badań klinicznych
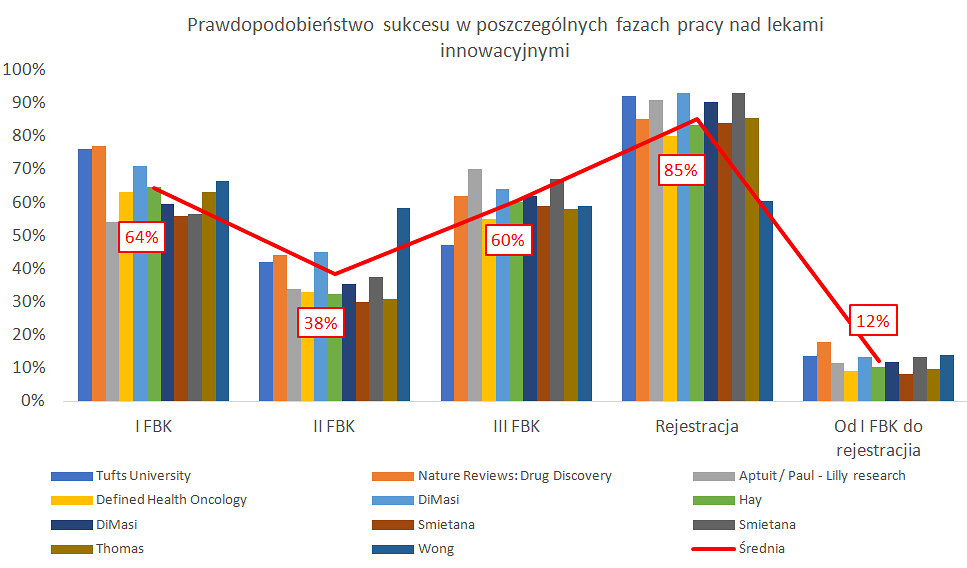
Kliknij, aby powiększyć.
Pierwszy z nich pokazuje średnie prawdopodobieństwo (bazujące na przebadaniu prawie 10 tys. projektów w latach 2006-2015) przejścia przez kolejne fazy badań klinicznych (63,2 proc., 30,7 proc., 58,1 proc. oraz 85,3 proc.). Slajd nr 2 to pozbierane przeze mnie wyniki różnych badań przeprowadzanych w różnych okresach i też ich uśrednienie.
Jak należy czytać te slajdy? Pierwsze cztery pozycje na linii X reprezentują pojedyncze prawdopodobieństwo przejścia leku do kolejnej fazy (z pierwszej fazy badań do drugiej, z drugiej do trzeciej, z trzeciej do złożenia wniosku o zatwierdzenia i w końcu uzyskanie pełnego dopuszczenia do obrotu). W efekcie końcowym otrzymujemy skumulowane prawdopodobieństwo zatwierdzenia leku, który wszedł do fazy I BK na to że uzyska status stanie leku dopuszczonego do obrotu. W pierwszym wykresie wynosi ona 9,5 proc., natomiast w drugim około 12 procent. Wartość ta matematycznie pochodzi z pomnożenia prawdopodobieństwa kolejnych faz przez siebie.
Powyższe wykresy pokazują, że statystycznie jeśli mamy 10 cząsteczek, które właśnie wchodzą do I fazy BK, to tylko jedna z nich zostanie lekiem. Dodatkowo jak widać na obydwu wykresach najtrudniej jest przejść drugą fazę. O powodach już pisałem, ale przypomnę, że jest to de facto pierwszy etap gdzie testuje się rzeczywistą skuteczność tego leku na ludziach. Po drugie, to też biznes – tutaj trzeba podjąć „męską decyzję” czy warto kontynuować projekt i ponosić koszty trzeciej fazy (kwoty do zainwestowania są zazwyczaj wysokie ze względu na liczbę uczestników, czas trwania, a także często konieczność zapłaty kolejnego kamienia milowego). Przecież może się okazać, że lek co prawda działa, ale wcale nie jest bardziej skuteczny niż te, które już funkcjonują na rynku, albo potencjał rynku jest już w danej chwili nie tak atrakcyjny jak w momencie rozpoczęcia badań nad takim lekiem.
Nie procenty są tutaj najbardziej istotne, ponieważ są one pewną statystyczną miarą. Jak wiadomo średnio ja i mój pies mamy 3 nogi, tylko, że niewiele z tego wynika. Konkretny lek, opracowujący go zespół i specyficzne uwarunkowania rynkowe w danym momencie mogą powodować, że prawdopodobieństwo danego projektu będzie istotnie różne od tych podanych powyżej. Zresztą mamy tutaj pewną cechę odróżniającą biznes biotechnologiczny od innych. Mówiąc najprościej jak się da, lek na końcu jest skuteczny lub nie jest skuteczny w leczeniu. Nawet jeśli jest skuteczny, może, ale nie musi, zostać zatwierdzony przez organy regulacyjne. Przed dopuszczeniem leki przechodzą ustrukturyzowany proces, podczas którego w dowolnym momencie mogą zawieść – a gdy zawiodą, proces ten jest często nieodwracalny. Oznacza to inny profil ryzyka niż większość innych firm, w których rozkład wyników jest jednak zazwyczaj mniej binarny.
Prawdopodobieństwo sukcesu badania klinicznego ma kluczowe znaczenie zarówno dla spółek biotechnologicznych jak i inwestorów do oceny przy podejmowaniu decyzji naukowych jak i biznesowych. Stąd też zachęcam do własnych poszukiwań różnego typu badań dostępnych w sieci. Jest to o tyle istotne, że w zależności np. typu choroby, bądź rodzaju leku, czy ścieżki zatwierdzania leku, statystyczne dane mogą się istotnie różnić. Proszę zresztą spojrzeć na kolejny slajd z wyżej przytoczonego badania.
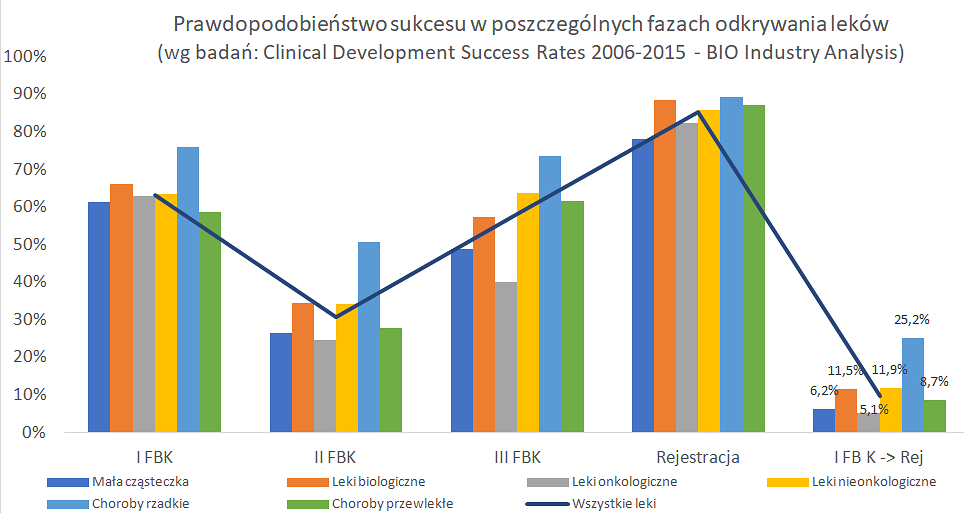
Kliknij, aby powiekszyc.
Powyższy wykres pokazuje prawdopodobieństwa sukcesu dla różnego typu leków (mała cząsteczka vs leki biologiczne) oraz typów chorób (leki onkologiczne vs leki nieonkologiczne oraz dla leków chorób rzadkich i chorób przewlekłych). Są one przedstawione na wykresie w kolumnach, natomiast wykres liniowy prezentuje dane dla całości zbadanej populacji. Jak widać, mogą występować dość istotne różnice w prawdopodobieństwie sukcesu w poszczególnych przypadkach.
Pierwsze porównanie dotyczy małych cząsteczek (NME- new molecular entity) z lekami biologicznymi, gdzie widać prawie dwukrotnie wyższe prawdopodobieństwo (11,5 proc. vs 6,2 proc.) sukcesu na rzecz leków biologicznych. Gdy porównamy leki onkologiczne z wszystkim pozostałymi, to tutaj też jasno widać dość niskie szanse leków onkologicznych – 5,1 proc. vs 11,9 proc. Jak widać skuteczny lek onkologiczny jest trudniej wynaleźć niż inne, co biorąc pod uwagę liczbę osób umierających rocznie na nowotwory wcale nie dziwi. Przecież wciąż niestety większość problemów nowotworowych nie została skutecznie rozwiązana. W wypadku onkologii trzeba również pamiętać o tym, że zaciera się trochę różnica między I a II fazą BK ze względu na testowanie od razu na pacjentach, a nie zdrowych ochotnikach. Leki onkologiczne również można podzielić na dwa (a tak naprawdę zapewne na więcej) różne typy: guzy lite (solid tumor) oraz nowotwory hematologiczne (hematological cancers). Guzy lite mają statystycznie dwukrotnie mniejszą szansę (4 vs 8,1 proc.) na przejście wszystkich faz badań klinicznych i zatwierdzenie.
Spójrzmy również na choroby rzadkie/sieroce (rare disease/orphan) oraz choroby przewlekłe o dużej częstości występowania (high prevalence chronic diseases). O ile te drugie mniej więcej sytuują się w średniej to choroby sieroce jak widać mają prawdopodobieństwo ponad 2,5 razy wyższe niż średnia (25,2 proc. vs 9,6 proc.), co też warto mieć na uwadze.
Mam nadzieję, że zauważyli Państwo pewne uproszczenie zastosowane w powyższych danych, które jest dość istotne i ważne. Otóż wszystkie te prawdopodobieństwa nie zawierają fazy odkrycia ani fazy badań przedklinicznych! Niestety, w szczególności w tym pierwszym wypadku zebranie takich wiarygodnych danych jest mocno utrudnione. Nie muszą być one raportowane do regulatorów, a spora część spółek (zespołów badawczych) na tym etapie prac nie jest notowana na giełdzie, co pociąga za sobą brak wymogów raportowania. A jak wiadomo sukcesami chwalimy się chętnie, ale porażkami już niekoniecznie. Tym niemniej w literaturze przyjęło się zakładać, że prawdopodobieństwo w fazie odkrywania to około 50 proc., natomiast w fazie przedklinicznej ok. 2/3. W efekcie gdy policzymy całkowitą szansę (9,6 proc. sukcesu od I fazy do zatwierdzenia) to spadnie ona do 3,4 proc.
Szczerze zachęcam do zapoznania się z w/w lub innymi raportami i ich dogłębnego przestudiowania. Warto o nich pamiętać, w szczególności gdy znajdziecie się Państwo akurat na prezentacji spółki omawiającej np. ekscytujące slajdy o potencjale rynku, liczbie chorób onkologicznych czy wartości biodollar value z kontraktów. To wszystko prawda, ale jak widać nie bierze się to znikąd – po prostu prawdopodobieństwo wynalezienia skutecznej innowacyjnej terapii jest dość niskie i warto mieć to z tyłu głowy, zwłaszcza przy podejmowaniu decyzji inwestycyjnych.
Zobacz także:
>> Biotechnologia na giełdzie. Czy drobni inwestorzy mają tu szansę na wielkie zyski?
>> Biotechnologia na giełdzie. Jak giełdowe spółki zarabiają na lekach i patentach
>> Wskaźniki i wyceny spółek biotechnologicznych na GPW
>> Wskaźniki i wyceny giełdowych producentów leków